

«Un tempo considerata una tecnologia futuristica nel distante orizzonte, la stampa 3D dei dispositivi medici, dei medicamenti e dei tessuti umani sta diventando rapidamente una promettente realtà»: l’affermazione è di Scott Gottlieb, il Commissioner della Food and Drug Administration (Fda).
La nuova linea guida marca solo il primo passo dell’approccio regolatorio a questa nuova frontiera tecnologica, ed è la stessa Fda a sottolineare come essa vada intesa come un documento “leap-frog”, ossia un ponte provvisorio destinato ad aggiornarsi nel tempo come conseguenza dell’evolversi delle tecnologie di manifattura additiva.
Tra le priorità poste in tal senso da Scott Gottlieb vi è l’approfondimento della messa in opera di processi di stampa 3D all’interno delle strutture “non tradizionali”, come possono essere i centri di ricerca in ambito universitario od ospedaliero o le stesse sale operatorie in cui venga effettuata la stampa “on-demand” in tempo reale di dispositivi su misura del paziente che giace sul tavolo operatorio.
Al momento restano inoltre esclusi dal campo di applicazione della nuova linea guida i prodotti stampati 3D che contengono cellule, tessuti o altre entità biologiche (prodotti che sono oggi normati alla stregua delle terapie avanzate).
Le classi dei dispositivi
La linea guida affronta gli aspetti prettamente tecnici della produzione additiva dei dispositivi medici, e deve essere integrata all’interno di tutta la già esistente normativa sulla classificazione e produzione dei medical device. L’approccio all’utilizzo di tecnologie di manifattura additiva per Fda deve essere declinato secondo una doppia prospettiva: da un lato, l’attenzione agli aspetti di progettazione e produzione (approfonditi nella prima sezione della linea guida, dedicata alle “Design and manufacturing considerations"), dall’altro l’approfondimento delle procedure di validazione e testing dei dispositivi così prodotti, i cui risultati devono essere inclusi nel dossier regolatorio (oggetto della seconda parte del documento, “Device testing considerations”) (La struttura della nuova linea guida Fda).
In termini generali, i dispositivi di classe II e III e alcuni dispositivi di classe I devono seguire procedure di sviluppo tali da assicurare anche la rispondenza a quanto disposto dal 21 CFR 820.30 “Design controls”.
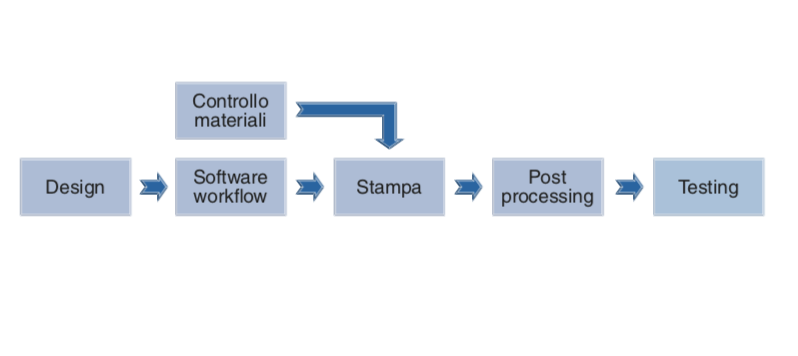
La manifattura additiva utilizza una grande varietà di tecnologie di stampa e di materiali di partenza, la cui scelta iniziale influenza profondamente il risultato ottenuto. Il cammino prescelto per lo sviluppo di un nuovo dispositivo stampato 3D può venire riassunto in un diagramma di flusso che evidenzi gli elementi chiave dell’intero ciclo produttivo.
La linea guida specifica anche che ogni stadio critico può essere ulteriormente dettagliato attraverso un sommario delle sue caratteristiche e richiede il monitoraggio continuo dei parametri di processi validati. Per ogni stadio, inoltre, devono essere identificati i relativi parametri e le specifiche di output, compresa la valutazione di come i parametri s’influenzano a vicenda e impattano sulle caratteristiche e la funzione del prodotto finale. Queste attività sono propedeutiche a garantire un’approfondita conoscenza da parte del fabbricante di ogni singolo passaggio della produzione e dei rischi ad esso associati (che devono sempre essere documentati insieme alle relative misure di mitigazione), indispensabile per poter individuare in modo tempestivo la root cause di eventuali difetti di produzione.
La struttura della nuova linea guida Fda
La linea guida Fda sulla manifattura additiva dei dispostivi medici è suddivisa in due grandi sezioni:
Design and manufacturing considerations: copre i requisiti tecnici che devono essere presi in considerazione per dimostrare la compliance del Sistema Qualità per la produzione del dispositivo medico. I requisiti devono essere determinati in base alla classificazione regolatoria del dispositivo e alla normativa ad esso applicabile.
Device testing considerations: descrive la documentazione regolatoria da presentare per le diverse tipologie di procedura autorizzativa. Vengono considerate le seguenti procedure: premarket notification (510(k)), premarket approval (Pma), humanitarian device exemption (Hde), de novo request e investigational device exemption (Ide).
La stampa 3D di dispositivi medici è tipicamente utilizzata per applicazioni su misura del singolo paziente, ad esempio per la produzione di protesi impiantabili in campo ortopedico o in neurochirurgia. La linea guida dell’Fda dedica una particolare attenzione alla progettazione dei diversi tipi di device – in serie o su misura – e, nel secondo caso, al ruolo svolto dai sanitari dei centri clinici o delle parti terze che prendano parte al processo. I fattori critici da considerare a questo riguardo comprendono l’effetto delle tecniche di imaging utilizzate per la diagnosi e la conseguente progettazione del dispositivo, l’alterazione dei parametri di un dispositivo standard per adattarlo al singolo paziente, la necessità di gestire file complessi che riproducono i parametri anatomici del paziente e i conseguenti aspetti di cybersicurezza e protezione dei dati personali. All’interno della prima parte della linea guida vengono anche discusse le caratteristiche del software di stampa e i controlli sui materiali di partenza, nonché i controlli post-processo e la validazione dell’intero iter produttivo (I passaggi della manifattura additiva dei dispositivi medici).
Documentare per l'approvazione regolatoria
Le informazioni che entrano a far parte dei dossier registrativi sono diverse a seconda della classificazione del dispositivo e devono essere adeguatamente validate e supportate dalle attività di testing descritte dalla seconda parte della linea guida. Tra le caratteristiche che non possono mancare vi sono le varie taglie in cui viene prodotto il dispositivo o l’identificazione delle dimensioni critiche che possono venire modificate per produrre dispositivi su misura.
Un aspetto centrale delle procedure di validazione è rappresentato dal fatto che per il momento sono disponibili ancora pochi test sul prodotto finito che recepiscano le specificità tipiche del processo di manifattura additiva, che può conferire caratteristiche peculiari al prodotto stesso rispetto ai metodi tradizionali di produzione.
I test per valutare le performance del prodotto sono per lo più ancora quelli per i dispositivi prodotti con tecnologie tradizionali, e devono essere condotti – dopo aver completato tutte le fasi di post-produzione, pulizia e sterilizzazione – in confronto a un worst-case-device la cui scelta deve essere sempre motivata. Tra i parametri critici da prendere in considerazione per la validazione dei test meccanici, vi è l’effetto sulle caratteristiche e la funzionalità del prodotto finito indotto dall’orientamento del processo di stampa e dal posizionamento del pezzo all’interno dello “spazio costruttivo” della stampante.
La peculiarità del processo di stampa additiva è data dalla capacità dei diversi strati di materiale che via via si aggiungono uno sull’altro di formare un unicum che dà vita al prodotto finito; la richiesta della linea guida a questo riguardo è di caratterizzare sempre le forze inter-strato di adesione e coesione, in quanto esse sono fondamentali per assicurare l’integrità del manufatto finale. Richieste ulteriori riguardano i materiali metallici o ceramici, per i quali è richiesta anche una valutazione della microstruttura, e quelli cristallini, per i quali va determinata la cristallinità e la morfologia dei cristalli, mentre per gli idrogel la linea guida chiede di determinare la percentuale di acqua di swelling.
La validazione dei materiali per la stampa additiva, che sono oggetto di profonde modificazioni nel corso del processo produttivo, richiede di fornire informazioni aggiuntive sulle modifiche chimiche che possono intercorrere in tal sede qualora esso alteri in modo inatteso i materiali e, di conseguenza, la loro biocompatibilità. Quest’ultima va valutata sul prodotto finito secondo lo standard ISO 10993. Ai fabbricanti potrebbero anche venire richieste informazioni aggiuntive sui rischi per la salute legate all’uso di polimeri come materia prima per la stampa. L’ente regolatorio americano specifica come l’approvazione del materiale prescelto per la stampa additiva non sia un processo a sé stante valido per tutti i dispositivi, ma vada piuttosto a collocarsi all’interno del processo di approvazione del singolo dispositivo medico e in relazione alla sua specifica destinazione d’uso. La procedura 510(k) può essere utilizzata anche per la notifica di dispositivi prodotti con materiali nuovi (senza bisogno di attivare la più rigorosa procedura di premarket review), qualora il materiale non presenti problemi di sicurezza ed efficacia e quando il dossier dimostri che esso è almeno ugualmente sicuro ed efficace rispetto a materiali equivalenti già presenti sul mercato.
Il processo produttivo non finisce con la stampa pura e semplice del pezzo. Sono infatti necessari processi di post-produzione per rimuovere i residui di materiali e le parti necessarie in fase di stampa (es. per sostenere il pezzo all’interno della camera della stampante) ma non necessarie alla funzionalità del prodotto finale: tutti aspetti che vanno opportunamente approfonditi e documentati, e che possono risultare difficili da gestire soprattutto nel caso di manufatti caratterizzati da una geometria complessa, che può avere un impatto rilevante anche sulle procedure di sterilizzazione prescelte. I test di degradazione in vitro sono richiesti nel caso di prodotti finiti stampati con materiali assorbenti.
Sicurezza ed efficacia
Scott Gottlieb prevede, nell’editoriale che ha accompagnato la pubblicazione della nuova linea guida, che le tecnologie di stampa 3D presto permetteranno il trattamento dei pazienti ustionati con impianti tissutali delle proprie stesse cellule stampate in 3D; un’altra prospettiva futura delineata dal Commissioner di Fda è la possibilità di riprodurre repliche di organi per i trapianti. Ma per poter giungere a queste applicazioni avanzate, molto più complesse da realizzare della stampa, ad esempio, di uno scaffold protettivo per le fratture o di una protesi, è necessario poter sviluppare tecnologie sempre più innovative e realmente “trasformative” per la pratica medica, che poggino su un robusto processo di sviluppo e su solide fondamenta regolatorie al fine di assicurare ai pazienti la sicurezza dei dispositivi, la loro efficacia e la possibilità di accedere senza difficoltà alle innovazioni tecnologiche.
Da questo punto di vista, gli Stati Uniti stanno al momento facendo da apripista per quanto riguarda l’implementazione delle tecnologie di manifattura additiva: da oltreoceano viene anche la prima approvazione di un farmaco stampato in 3D, il prodotto di Aprecia Pharmaceuticals per il trattamento dell’epilessia (ne abbiamo parlato sul numero di maggio 2016 di NCF). Il Center for drug evaluation and research (Cder), è l’ente dell’Fda preposto allo studio dell’impatto delle tecnologie di stampa additiva sulla produzione dei medicinali, mentre il Center for devices and radiological health (Cdrh) supporta tutte le attività legate alla produzione, repackaging, realelling e importazione dei dispositivi medici negli Stati Uniti. Entrambi sono dotati di certi di stampa 3D allo stato dell’arte in cui testare i diversi approcci alla manifattura additiva, e hanno contribuito con la propria esperienza alla messa a punto dei requisiti che sono entrati a far parte della nuova linea guida dell’ente regolatorio americano.
I passaggi della stampa 3D dei dispositivi medici
Il numero di passaggi necessari alla produzione di un singolo dispositivo è variabile, e dipende dalla complessità del dispositivo stesso. Il processo tipico delineato da Fda prevede i seguenti stadi:
- Device design: il dispositivo è progettato e validato mediante modelli digitali di taglia pre-specificata, o modelli digitali su misura delle dimensioni anatomiche del paziente;
- Software workflow: il progetto digitale è convertito in un file eseguibile dalla stampante 3D, che spesso suddivide il progetto in strati successivi e include in esso anche il materiale aggiuntivo di supporto per la stampa. Il software indica alla stampante come posizionare il pezzo sulla piattaforma di stampa. La stampante può richiedere adattamenti (es. variazioni di materiali, tipo di progetto, destinazione d’uso) per la produzione di diversi tipi di prodotto;
- Material controls: i materiali usati per la produzione devono essere di elevata qualità e rispondenti alle specifiche. A tal fine, devono essere previsti accordi tra fornitore, acquirente e utilizzatore finale delle materie prime circa i requisiti, le procedure e i controlli sui materiali che devono essere effettuati su ogni lotto di materiale;
- Printing: l’oggetto è stampato secondo le specifiche progettuali previste dal file di stampa;
- Post-Processing: possono essere necessari uno o più passaggi successivi alla stampa, per es. la rimozione dei residui di materiale, il raffreddamento controllato (annealing) e/o procedure di drilling, taglio, lucidatura e sterilizzazione.
- Process validation and verification: Alcuni dispositivi o componenti caratteristiche (come gli elementi geometrici) possono essere testate singolarmente per verificare la rispondenza alle specifiche e il corretto funzionamento. Nel caso di caratteristiche funzionali come la forza meccanica, la cui validazione sul singolo pezzo è difficile in quanto potrebbe portare a distruzione dello stesso, esse devono essere oggetto di una validazione del processo preliminare alla produzione. In questo modo si assicura che il processo produttivo dia luogo a prodotti rispondenti alle specifiche definite, previo monitoraggio e controllo degli specifici parametri di processo;
- Testing: I metodi e i risultati dei test devono essere sottoposti a Fda per dimostrare la rispondenza ai requisiti regolatori e la sicurezza ed efficacia del dispositivo rispetto all’uso previsto. Ad ogni dispositivo corrisponde un insieme specifico di test, da individuare sulla base delle linee guida Fda, degli standard internazionali o dei processi di controllo interni. I dispositivi stampati in 3D sono in generale soggetti agli stessi requisiti regolatori di quelli prodotti con metodi tradizionali.




: “Il 2026 è l’anno in cui è diventata reale”")




{kind=link}